FastQ screen is a tool from Babraham Bionformatics group and is found here. FastQ screen allows user to screen for contaminations (artifacts) in fastq files. They may be from other organisms of your interest or from vector or primers.
FastQ screen program needs following information to work:
- Source files (.fastq, .fq and gzipped files)
- Indexed reference genome and genomes you want to look for (for eg. mouse, pig etc)
- Indexed contaminant sequences (for eg primer sequences or vector sequences)
- Aligners- currently it supports 3 aligners: bwa, bowtie and bowtie2.
- Configuration file with reference genome index and any other indexes (primer, vector)
Since it requires aligner, reference and other fasta files must be indexed with same aligner that would be used in FastQ screen execution. For eg. if you are using bowtie2 aligner in executing FastQ screen, you need to index your files (.fa) with bowtie2.
Let us work with an example. Download FastQ screen from here. Download the raw files from here. Let us work with one file only as an example. The fastq files align well with Chr22 and download chr22.fa (hg38/GRCh38 is preferred) for indexing. When you index fasta files (with bowtie2) it would create index files with .bt2 extension.
Please keep source (.fa) and index files in individual folders as index files will be too many (5 index files for chr22). For eg. Keep reference chr22 fasta file in a separate folder and primer sequence (.fa) in a separate folder. Then index them.
$ bowtie2-build -f hg38.chr22.fa hg38.chr22hg38.chr22.fa - Reference fasta
hg38.chr22 - Base name for index files
Step 2: Index primer sequences. Download Illumina primers sequences from here and Index them.
$ bowtie2-build -f gsaf_illumina_adapters.fasta illumina
gsaf_illumina_adapters.fasta - Illumina Adapter sequences
$ bowtie2-build -f gsaf_illumina_adapters.fasta illumina
gsaf_illumina_adapters.fasta - Illumina Adapter sequences
illumina - Base name for index files
When you download and gunzip FastQ screen source, you would be provided with an example conf. Try to reuse this configuration file. Configuration file used for this note is as below:
$ cat fastq_screen.conf
############
## Threads #
############
THREADS 6
##############
THREADS 6
##############
## DATABASES #
##############
DATABASE Human /home/<user>/reference/hg38/hg38.chr22
DATABASE Adapters /home/<user>/Desktop
===================================================DATABASE Human /home/<user>/reference/hg38/hg38.chr22
DATABASE Adapters /home/<user>/Desktop
User can add many databases to screen sequences against, as per the mentioned format:
DATABASE Human /home/<user>/reference/hg38/hg38.chr22
(Database, Organsim name, Location of index files and base name of index: in this case: hg38.chr22 -- refer to your bowtie2 index basename)
DATABASE Adapters /home/<user>/Desktop(Database, General name, Location of Adapters fasta file. Index files must be located in the same folder)
Note that for reference, you need to furnish location and basename of index and for primers, vectors you need to furnish location of fasta file it self.
Final command to execute FastQ screen is:
$ ./fastq_screen -conf fastq_screen.conf /home/<>/raw_data/hcc1395_normal_rep1_r1.fastq.gz Please note that for this note, I have used only human sequence (chr22) and primer sequence only. Output would be in 3 formats: html, png and txt.
It would be as follows:
png
png
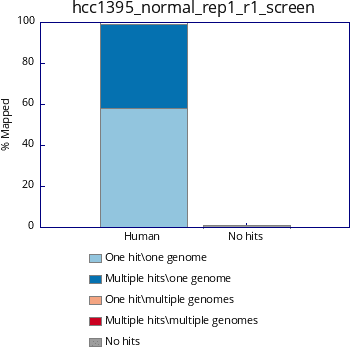
Text:
$ cat hcc1395_normal_rep1_r1_screen.txt
#Fastq_screen version: 0.11.1 #Aligner: bowtie2 #Reads in subset: 100000
Genome #Reads_processed #Unmapped %Unmapped #One_hit_one_genome %One_hit_one_genome #Multiple_hits_one_genome %Multiple_hits_one_genome #One_hit_multiple_genomes %One_hit_multiple_genomes Multiple_hits_multiple_genomes %Multiple_hits_multiple_genomes
Human 110652 1306 1.18 64380 58.18 44966 40.64 0 0.00 0 0.00
%Hit_no_genomes: 1.18
html (screenshot):
#Fastq_screen version: 0.11.1 #Aligner: bowtie2 #Reads in subset: 100000
Genome #Reads_processed #Unmapped %Unmapped #One_hit_one_genome %One_hit_one_genome #Multiple_hits_one_genome %Multiple_hits_one_genome #One_hit_multiple_genomes %One_hit_multiple_genomes Multiple_hits_multiple_genomes %Multiple_hits_multiple_genomes
Human 110652 1306 1.18 64380 58.18 44966 40.64 0 0.00 0 0.00
%Hit_no_genomes: 1.18
html (screenshot):
