Transcriptomic data's final goal is to identify the differential entities (genes/transcripts/exons/isoforms) between or among conditions with statistical significance. Classical way of this in NGS is to use splice aware aligners, transcript reconstitution (via exons), followed by quantification and then differential analysis. Next generation tools in transcriptomic data analysis skip alignment totally, use other approaches to quantify the transcripts. Some of these are claim to be faster than traditional aligners. There are three such popular pseudo aligners/quantification tools: sailfish, Kallisto and salmon. These tools do not produce bam or sam. They produce quantification files direct or use existing bams from traditional aligners for quantification.
In today's note, we will try salmon and subsequent differential analysis tools.

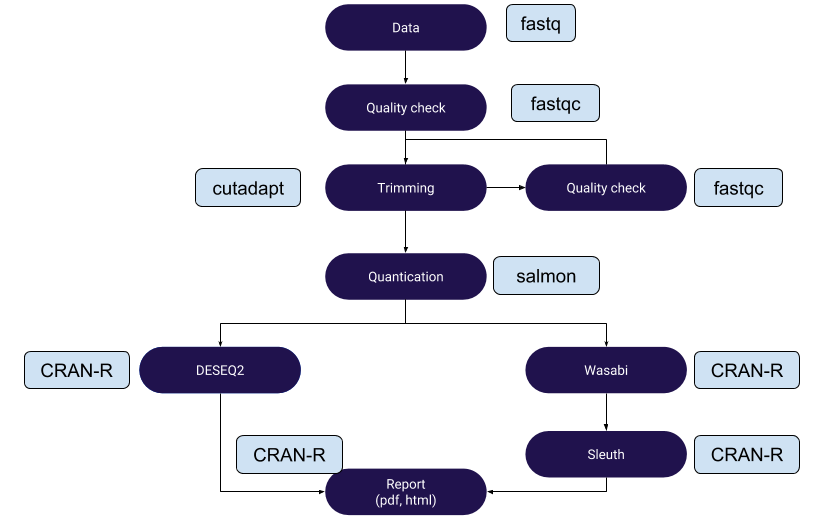
Above work flow is written in Snakemake and is available on my Github repo. Data can be downloaded from here from Griffith lab tutorials. There are certain things that need attention:
1. Salmon needs fasta sequence for transcriptome only, not genome sequence. So one needs sequences for transcripts only. In addition it also needs transcript-gene mapping file (as tsv). For chromosome 22, I made it available here. Sequence is zipped and mapping file is provided as tsv. Note that the information is downloaded from UCSC using table browser and redundant entries are removed.
1. Salmon needs fasta sequence for transcriptome only, not genome sequence. So one needs sequences for transcripts only. In addition it also needs transcript-gene mapping file (as tsv). For chromosome 22, I made it available here. Sequence is zipped and mapping file is provided as tsv. Note that the information is downloaded from UCSC using table browser and redundant entries are removed.
2. For report generation in R, you would need a library called "regionReport". This in turn needs a latex to pdf converter. Do not install texlive from repositories. It causes lots of headache. Instead, install tinytex package, available in CRAN-R repos. Once installed, run "install_tinytex" to install necessary packages. This in turn install, necessary binaries/functions in a bin folder in your home folder.
3. Use bootstrap option (while executing Salmon) if you want to use wasabi-sleuth work flow. Without quantification bootstrap option, sleuth will not work although wasabi package works fine.
Here is the github (written by me) link to Snakemake scripts for Salmon-DESEQ2 and Salmon-Wasabi-Sleuth work flow. Make sure that your raw data, reference and output folders are edited proper in the scripts.
Instructions to run the script:
- Download all the files from Github. Keep it any where in you like. But keep all of them in the same folder.
- Create a directory of your project (for eg project_a)
- Move config.yaml (in github directory) to the folder (for eg. project_a)
- Create a new directory called "reference" in the new folder (for eg. project_a/reference)
- Now move to reference directory. Download fasta file and transcript to gene map files from here.
- Make a new directory by name "raw_data" and keep the paired fastq files in the directory, downloaded from griffith lab.
- Now there are two directories, where the snakemake files are stored. There is another directory (project_a) with raw_data, config.yaml and reference files.
- Execute the snakemake workflow by following command:
$ snakemake -j 4 -s Snakefile --directory ../project_a