Salmon and Salmon output downstream analysis requires two files as reference:
User can use either entire transcriptome or part transcriptome. NCBI provides sequences files for full transcriptome. For partial sequence, I would suggest UCSC table browser.- Transcriptome sequence (not genome sequence) in fasta (fa) or fasta.gz (fa.gz) format
- Mapping between Transcripts and corresponding genes
Now let us first deal with NCBI full transcriptome sequence:
- File name is GRCh38_latest_rna.fna.gz and can be downloaded from here: ftp://ftp.ncbi.nlm.nih.gov/refseq/H_sapiens/annotation/GRCh38_latest/refseq_identifiers. This file would suffice for indexing (by Salmon) and subsequent quantification.
- Downstream tools like DESEQ2 and Wasabi/Sleuth need transcript to gene mapping file and they should match with those used in quantification. Following is the bash script that generates transcript to gene mapping using the fasta headers from GRCh38_latest_rna.fna.gz.
$ zgrep \> GRCh38_latest_rna.fna.gz | sed 's/),\strans.*//g' | sed 's/),\s.*RNA//g' | awk -v OFS="\t" -F " " '{print $1,$NF}' | awk '{gsub("[>,(,)]","",$0)}1' > GRCh38_t2g.tsv
====================
This tsv will be used in DESEQ2 object making and also in Sleuth object post Wasabi data transformation. Let us look at the output:
=====================
$ zgrep \> GRCh38_latest_rna.fna.gz | sed 's/),\strans.*//g' | sed 's/),\s.*RNA//g' | awk -v OFS="\t" -F " " '{print $1,$NF}' | awk '{gsub("[>,(,)]","",$0)}1' | head
NM_000014.5 A2M
NM_000015.2 NAT2
NM_000016.5 ACADM
NM_000017.3 ACADS
NM_000018.3 ACADVL
NM_000019.3 ACAT1
NM_000020.2 ACVRL1
NM_000021.3 PSEN1
NM_000022.3 ADA
NM_000023.3 SGCA
=====================
Apparently sleuth is sensitive to duplicate entries. So be careful about them. By the way, Salmon only takes NM entries from NCBI header. It doesn't use entire header. For eg. NCBI header is ">NM_000014.5 Homo sapiens alpha-2-macroglobulin (A2M), transcript variant 1, mRNA". Salmon uses only ">NM_000014.5" from the header.
Now let us have a look at partial transcriptome. To get partial transcriptome use UCSC table browser. Navigate to website, and follow the screenshots:
Choose the group (genes and Gene predictions), track (NCBI RefSeq), region (of interest) and output format.

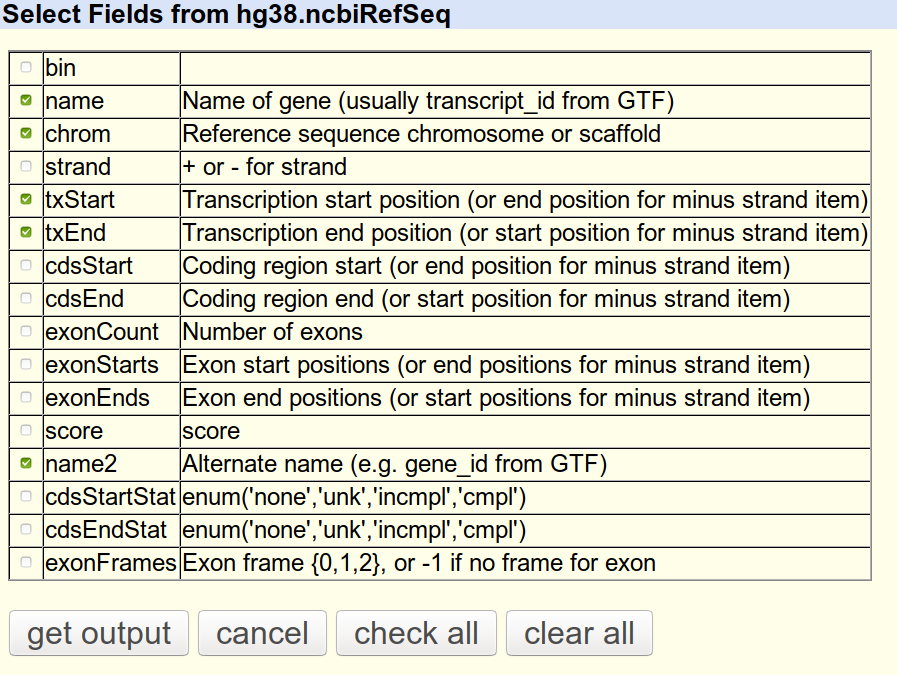
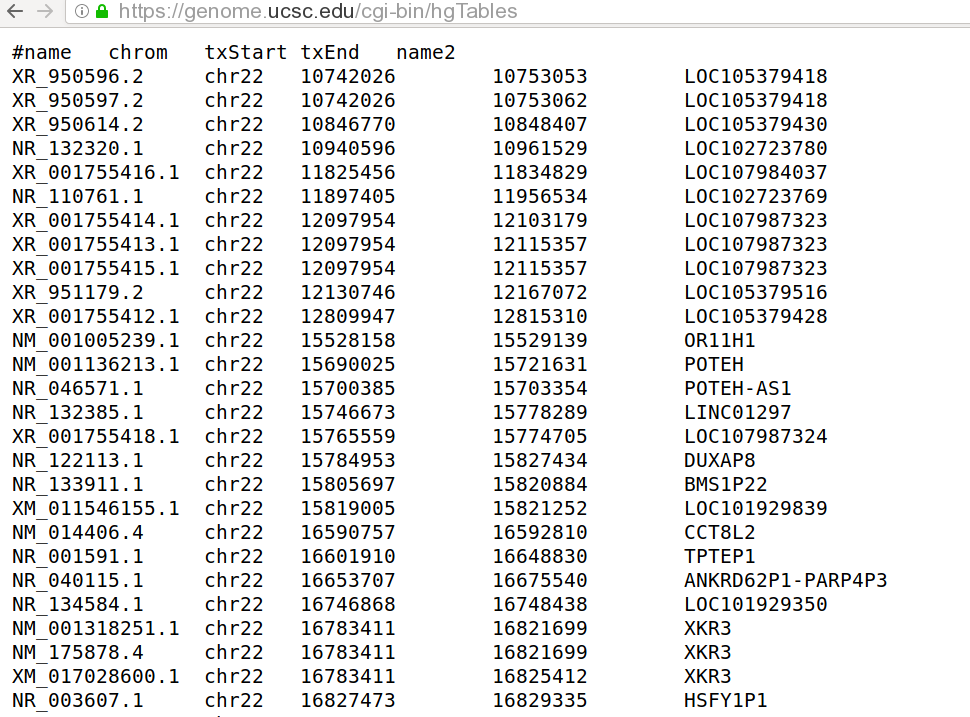
Now we need transcript, transcript coordinates and genes. Let us have a look at the downloaded file:
=================================
$ head chr22_ucsc_grch38_refseq.gtf
#name chrom strand txStart txEnd name2
NR_132320 chr22 - 10940596 10961529 FRG1FP
NR_110761 chr22 + 11897405 11956534 LOC102723769
NM_001005239 chr22 + 15528158 15529139 OR11H1
NM_001136213 chr22 + 15690025 15721631 POTEH
NR_046571 chr22 - 15700385 15703354 POTEH-AS1
NR_132385 chr22 + 15746673 15778289 LINC01297
NR_122113 chr22 + 15784953 15827434 DUXAP8
NR_073459 chr22 - 15805697 15815897 BMS1P18
NR_073460 chr22 - 15805697 15815897 BMS1P17
===================================
We will deduplicate transcript names (as it is necessary for Sleuth workflow) and then extract the chromosome name, coordinates and transcript names:
=========================================
$ awk 'BEGIN {!/#/} !a[$1]++' chr22_ucsc_grch38_refseq.gtf | awk -v OFS="\t" '!/#/ {print $2,$4,$5,$1}' > chr22_ucsc_grch38_refseq.bed
=========================================
About code:
awk 'BEGIN {!/#/} !a[$1]++' chr22_ucsc_grch38_refseq.gtf - De dups all lines that doesn't start with # and dedup is based on column 1.Rest of the code reorders the columns.
Let us look at the bed first:
==========================
$ head chr22_ucsc_grch38_refseq.bed
chr22 10940596 10961529 NR_132320
chr22 11897405 11956534 NR_110761
chr22 15528158 15529139 NM_001005239
chr22 15690025 15721631 NM_001136213
chr22 15700385 15703354 NR_046571
chr22 15746673 15778289 NR_132385
chr22 15784953 15827434 NR_122113
chr22 15805697 15815897 NR_073459
chr22 15805697 15815897 NR_073460
chr22 15805697 15820884 NR_133911
==========================
Let us use this information to extract sequences for transcripts:
To extract transcriptome sequence from genome sequence, we need bedtools installed on the machine. Bedtools provide getFasta function to extract sequence of interest if one provides reference sequence (in fasta or gzipped fasta format) and a bed file with coordinates and names. In current example, I used chr22 (genomic) sequence only. User can use full genome or regions of interest sequence. User can download the chromosome specific sequence (fa) from UCSC and ensembl. Please remember use the sequence and annotation from same source (NCBI, UCSC, ENSEMBL) through out the analysis. For eg, if you use ensembl sequence in alignment, it is better to use ensembl transcripts and ensembl genes for annotation. Same goes with NCBI and UCSC. I have used UCSC-Refseq for most of the work.
===============================
bedtools getfasta -name -fi hg38.chr22.fa -bed chr22_ucsc_grch38_refseq_dedup.bed > chr22_ucsc_grch38_refseq_dedup.fa
===============================
This would create fasta sequences only for transcripts and puts in output. This fasta you should use indexing for salmon. Please note that I took chr22 as an example only.
Now to extract transcript to gene mapping:
=====================
$ awk -v OFS="\t" '{print $1,$4}' chr22_ucsc_grch38_refseq_dedup.bed> t2g.tsv
$ head t2g.tsv
chr22 NM_000026
chr22 NM_000106
chr22 NM_000185
chr22 NM_000262
chr22 NM_000268
chr22 NM_000343
chr22 NM_000355
chr22 NM_000362
chr22 NM_000395
chr22 NM_000398
========================
This is the tsv file one should be using in downstream DESEQ2 and Wasabi-Sleuth workflow