One of the most used tasks in ngs analysis is to create a single reference file in either hgxx (for eg hg19, hg38) or b3x (b32, b35) format. For hg19, it is explained below. I downloaded hg19 reference files from UCSC goldenpath with name: hg19_gp_chromFa.tar.gz (current version is hg38 ). When I unzip the gzipped file, I get multiple fasta files. I got rid of chrun and hap files (for this example sake). I am left with only chr<number, X,Y, M>.fa files. These files are to be merged into one single reference file (hg19) either in bxx (eg. b32) or hgxx (hg18, hg19) format. Since they are from UCSC, all files start with "chr" word.
ls *.fa command in GNU-linux would output files in following order: chr10.fa chr14.fa chr18.fa chr21.fa chr4.fa chr8.fa chrY.fa chr11.fa chr15.fa chr19.fa chr22.fa chr5.fa chr9.fa chr12.fa chr16.fa chr1.fa chr2.fa chr6.fa chrM.fa chr13.fa chr17.fa chr20.fa chr3.fa chr7.fa chrX.fa.
This is not required order. Easiest way to get them sorted by human readable chromosome number is:
$ ls *.fa | sort -V | grep -i -v chrM
This command would remove chrM from the list and chromosomes are sorted in natural order. If one has to create a single reference files from chr*.fasta files, use following command (in bash shell):
Example: $ grep chr b32.fa and output is as show below:
 Please note that b3x style doesn't have "chr" appended to chromosomes. Names would be 1 to 22, X, Y, MT
Please note that b3x style doesn't have "chr" appended to chromosomes. Names would be 1 to 22, X, Y, MT
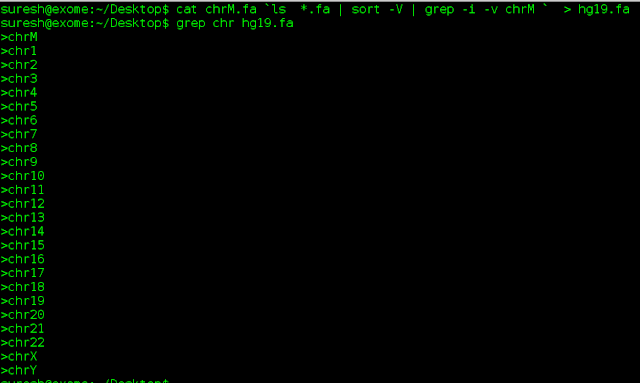
$ grep chr hg19.fa
ls *.fa command in GNU-linux would output files in following order: chr10.fa chr14.fa chr18.fa chr21.fa chr4.fa chr8.fa chrY.fa chr11.fa chr15.fa chr19.fa chr22.fa chr5.fa chr9.fa chr12.fa chr16.fa chr1.fa chr2.fa chr6.fa chrM.fa chr13.fa chr17.fa chr20.fa chr3.fa chr7.fa chrX.fa.
This is not required order. Easiest way to get them sorted by human readable chromosome number is:
$ ls *.fa | sort -V | grep -i -v chrM
This command would remove chrM from the list and chromosomes are sorted in natural order. If one has to create a single reference files from chr*.fasta files, use following command (in bash shell):
Creating b3x style reference:
$ cat `ls *.fa | sort -V | grep -i -v chrM ` chrM.fa > b32.fa
(Please note that this doesn't work in fish shell)
To check if chromosomes are ordered as expected, run following command:
$ grep chr <final>.faExample: $ grep chr b32.fa and output is as show below:

Creating hgxx style reference:
$ cat chrM.fa `ls *.fa | sort -V | grep -i -v chrM ` > hg19.fa
To view the order of chromosomes in fasta file,$ grep chr hg19.fa
