Files in fasta format are simplest formatted sequence files with a header in single line, followed by sequence it self, be it protein or nucleotide. There are several tools to parse fasta. Earlier notes from this blog displayed how to simulate sequences (DNA and protein). Note here explains how to extract sequences from file (based on their names) and write each sequence to a single file. For this we need two programs: Seqtk (available in ubuntu rep) and FaSplit (from kent resources : http://hgdownload.cse.ucsc.edu/admin/exe/).
After creating a file with random sequences, we may want to extract few sequences from this file, into another file. Example file can be downloaded from here. This example file has with 10 fasta sequences (a to j.fasta) with 10 bases(nt) each.
After creating a file with random sequences, we may want to extract few sequences from this file, into another file. Example file can be downloaded from here. This example file has with 10 fasta sequences (a to j.fasta) with 10 bases(nt) each.
 |
| 10 sequences in fasta format |
This note outlines how to extract selected sequences, extract all the sequences into individual files and also convert fasta file with two column file (name and sequences separated by a tab).
1) Extract sequences of interest
Logic: Extract the names of the sequence and create a list of wanted sequences. Pass this list to program to extract fasta sequences of interest into another fasta file.
Extract sequence names from the fasta file (without > symbol):
Command: grep ">" ds.seq.fa | cut -c2- >names (screenshot of names)

{kind=link}
2) Now that the file names are in stored in a file called names. (refer to screenshot above)
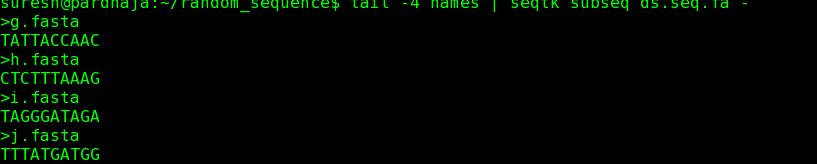
3) Now, let us say we want last 4 sequences from the original file , execute following command.
command: tail -4 names | seqtk subseq ds.seq.fa - >tail.fa.
After exeuction, a new file would be created by name "tail.fa" with last 4 fasta sequences in ds.seq.fa ( as listed in file name "names"). Refer to screen shot for extracted 4 sequences.
Now let us you would like to extract few sequences from the fasta file. Let us make a dummy fasta file with 3 sequences: a, b and c.
$ cat test.fa
>a
atgc
>b
ctga
>c
gtca
>a
atgc
>b
ctga
>c
gtca
Now let us say we would like to extract sequences b and c. Now what you have to do is make a text file. Text file should have ids (with > sign), one line per ID. Example text file for extracting sequences b and c look like this:
$ cat list
b
c
b
c
Now let us execute the command for desired sequences:
$ seqtk subseq test.fa list
>b
ctga
>c
gtca
>b
ctga
>c
gtca
2) Extract all the sequences and write into individual fasta files
Command: faSplit byname tnames.fa .
This command would extract sequences mentioned by their name (in the file tnames) into current directory (. denotes current directory). Program faSplit x86-64 linux binary can be downloaded from here (~1MB executable).
 |
| Before: Note that all the sequences are within a single file named "tnames.fa" |
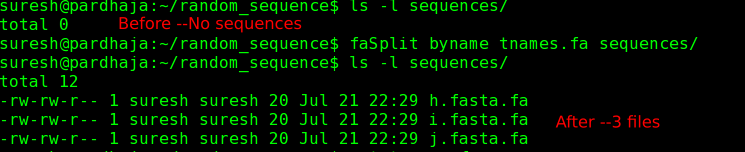 | ||||
| Before and After: In destination directory, there are no files before splitting. After splitting, 3 files are created afresh |
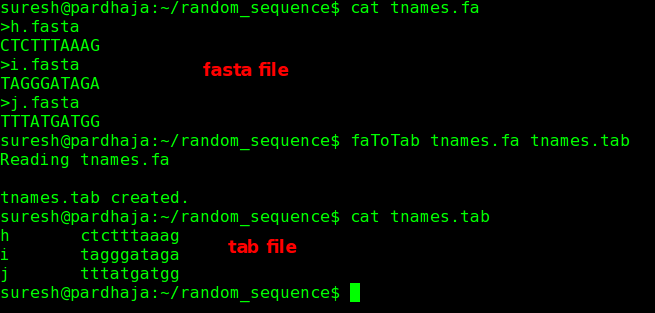
3) Create tab separated sequences from fasta file
Command: faToTab <input.fa> <output>
Example: faToTab tnames.fa tnames.tab
faToTab can be downloaded from here (~1 MB x86-64 GNU-linux executable)