RNA seq in general is used for quantification of expression. However, the same data can be used for variant identification and further annotation. There are pipelines that outline variant calling using RNAseq data including GATK best practices workflow for variant calling using RNA seq data. In addition, there are several NGS data analysis and annotation specific platforms are being available now a days like Gemini (for variant annotation). Few of the data analysis (pipeline) tools are Pegassus, Galaxy, Bpipe, Ruffus etc. A small comparison can be found here.
In this note, a small Bpipe script is included for calling variants. Workflow of the pipeline is explained below:

Bedtools (vcf intersection), Bwa (alignment and indexing), freebayes (variant calling), picardtools (deduplication), samtools (sorting and variant calling), vcflib (variant merging, sorting)
Following is the pipeline: Please replace with appropriate directories on your system
====================================================================
fastqc={
exec "fastqc ./data/reads.end1.fq -o ./fastqc"
exec "fastqc ./data/reads.end2.fq -o ./fastqc"
}
cutadapt={
exec "cutadapt -q 20 -o ./cutadapt/reads.end1.cutadapt.fq -p ./cutadapt/reads.end2.cutadapt.fq ./data/reads.end1.fq ./data/reads.end2.fq > ./cutadapt/cutadapt_qual.log"
}
indexref={
exec "bwa index ./data/chr20.fa"
}
align={
exec "bwa mem -t 4 -M ./data/chr20.fa ./cutadapt/reads.end1.cutadapt.fq ./cutadapt/reads.end2.cutadapt.fq > ./bwa/bwa.err > ./bwa/na12878.q20.cutadapt.sam"
}
sort={
exec "samtools view -bSu ./bwa/na12878.q20.cutadapt.sam | samtools sort - ./samtools/na12878.q20.cutadapt.sorted"
}
dedup={
exec "java -jar /opt/picard-tools-1.119/MarkDuplicates.jar METRICS_FILE=./picard/na12878.q20.cutadapt.sorted.dedup.metrics REMOVE_DUPLICATES=true ASSUME_SORTED=true VALIDATION_STRINGENCY=LENIENT I=./samtools/na12878.q20.cutadapt.sorted.bam O=./picard/na12878.q20.cutadapt.sorted.dedup.bam"
}
bamindex={
exec "samtools index ./picard/na12878.q20.cutadapt.sorted.dedup.bam"
}
samtools_vc = {
exec "samtools mpileup -uf ./data/chr20.fa ./picard/na12878.q20.cutadapt.sorted.dedup.bam | bcftools call -mv -Ov - > ./variantcalling/samtools.na12878.q20.cutadapt.sorted.dedup.vcf"
}
freebayes_vc={
exec "freebayes -f ./data/chr20.fa ./picard/na12878.q20.cutadapt.sorted.dedup.bam > ./variantcalling/freebayes.na12878.q20.cutadapt.sorted.dedup.vcf"
}
gatk_vc= {
exec "java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar -T HaplotypeCaller -R ./data/chr20.fa -I ./picard/na12878.q20.cutadapt.sorted.dedup.bam -L 20 --genotyping_mode DISCOVERY -stand_emit_conf 10 -stand_call_conf 30 -o ./variantcalling/gatk.na12878.q20.cutadapt.sorted.dedup.vcf"
}
mergevcf= {
exec "vcfcombine ./variantcalling/*.vcf > ./variantcalling/fb_sam.vcf"
}
uniquevcf= {
exec "vcfuniq ./variantcalling/fb_sam.vcf > ./variantcalling/fb_sam.uniq.vcf"
}
cvariants={
exec "bedtools intersect -a ./variantcalling/fb_sam.uniq.vcf -b ./data/hg19.genome.refseq.gtf -wa -wb > ./variantcalling/coding_variants.txt"
}
ncvariants={
exec "bedtools intersect -v -a ./variantcalling/fb_sam.uniq.vcf -b ./data/hg19.genome.refseq.gtf > ./variantcalling/noncoding_variants.txt"
}
Bpipe.run {
fastqc+cutadapt+indexref+align+sort+dedup+bamindex+samtools_vc+freebayes_vc+mergevcf+uniquevcf+cvariants+ncvariants
}
====================================================================
In this note, a small Bpipe script is included for calling variants. Workflow of the pipeline is explained below:
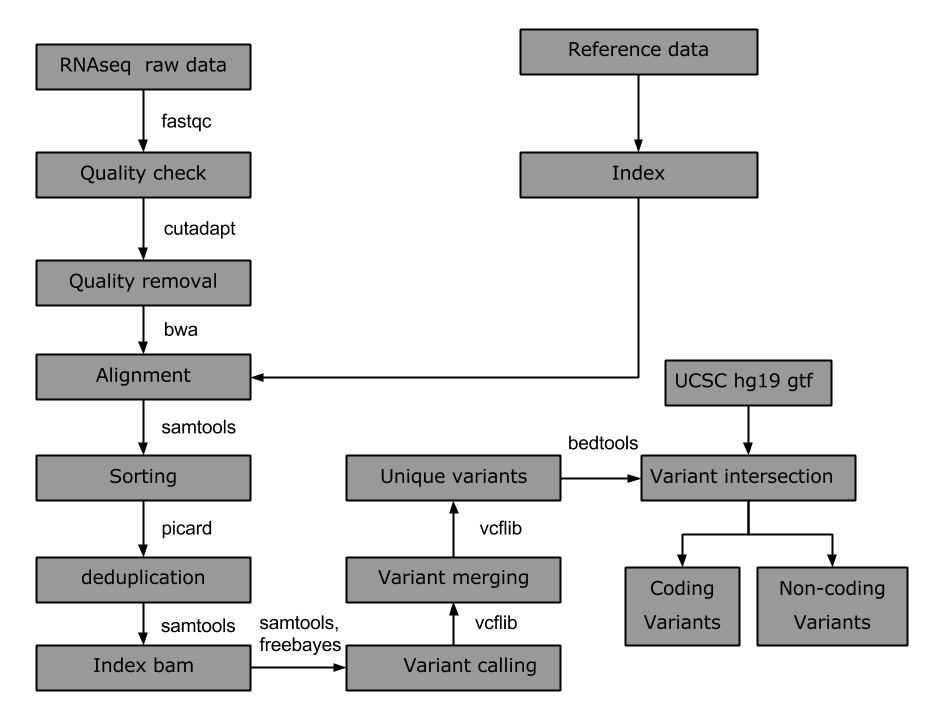
Tools used
Bedtools (vcf intersection), Bwa (alignment and indexing), freebayes (variant calling), picardtools (deduplication), samtools (sorting and variant calling), vcflib (variant merging, sorting)
Following is the pipeline: Please replace with appropriate directories on your system
====================================================================
fastqc={
exec "fastqc ./data/reads.end1.fq -o ./fastqc"
exec "fastqc ./data/reads.end2.fq -o ./fastqc"
}
cutadapt={
exec "cutadapt -q 20 -o ./cutadapt/reads.end1.cutadapt.fq -p ./cutadapt/reads.end2.cutadapt.fq ./data/reads.end1.fq ./data/reads.end2.fq > ./cutadapt/cutadapt_qual.log"
}
indexref={
exec "bwa index ./data/chr20.fa"
}
align={
exec "bwa mem -t 4 -M ./data/chr20.fa ./cutadapt/reads.end1.cutadapt.fq ./cutadapt/reads.end2.cutadapt.fq > ./bwa/bwa.err > ./bwa/na12878.q20.cutadapt.sam"
}
sort={
exec "samtools view -bSu ./bwa/na12878.q20.cutadapt.sam | samtools sort - ./samtools/na12878.q20.cutadapt.sorted"
}
dedup={
exec "java -jar /opt/picard-tools-1.119/MarkDuplicates.jar METRICS_FILE=./picard/na12878.q20.cutadapt.sorted.dedup.metrics REMOVE_DUPLICATES=true ASSUME_SORTED=true VALIDATION_STRINGENCY=LENIENT I=./samtools/na12878.q20.cutadapt.sorted.bam O=./picard/na12878.q20.cutadapt.sorted.dedup.bam"
}
bamindex={
exec "samtools index ./picard/na12878.q20.cutadapt.sorted.dedup.bam"
}
samtools_vc = {
exec "samtools mpileup -uf ./data/chr20.fa ./picard/na12878.q20.cutadapt.sorted.dedup.bam | bcftools call -mv -Ov - > ./variantcalling/samtools.na12878.q20.cutadapt.sorted.dedup.vcf"
}
freebayes_vc={
exec "freebayes -f ./data/chr20.fa ./picard/na12878.q20.cutadapt.sorted.dedup.bam > ./variantcalling/freebayes.na12878.q20.cutadapt.sorted.dedup.vcf"
}
gatk_vc= {
exec "java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar -T HaplotypeCaller -R ./data/chr20.fa -I ./picard/na12878.q20.cutadapt.sorted.dedup.bam -L 20 --genotyping_mode DISCOVERY -stand_emit_conf 10 -stand_call_conf 30 -o ./variantcalling/gatk.na12878.q20.cutadapt.sorted.dedup.vcf"
}
mergevcf= {
exec "vcfcombine ./variantcalling/*.vcf > ./variantcalling/fb_sam.vcf"
}
uniquevcf= {
exec "vcfuniq ./variantcalling/fb_sam.vcf > ./variantcalling/fb_sam.uniq.vcf"
}
cvariants={
exec "bedtools intersect -a ./variantcalling/fb_sam.uniq.vcf -b ./data/hg19.genome.refseq.gtf -wa -wb > ./variantcalling/coding_variants.txt"
}
ncvariants={
exec "bedtools intersect -v -a ./variantcalling/fb_sam.uniq.vcf -b ./data/hg19.genome.refseq.gtf > ./variantcalling/noncoding_variants.txt"
}
Bpipe.run {
fastqc+cutadapt+indexref+align+sort+dedup+bamindex+samtools_vc+freebayes_vc+mergevcf+uniquevcf+cvariants+ncvariants
}
====================================================================
Note: Yet to add annotations using VEP (ENSEMBL) and SNpeff. Please note that intersection using bedtools can also take bed files for annotation.