GATK, provided by Broad institute, has become defacto standard for calling variants in WES, WGS and targeted/panel based sequencing data for many labs across the world. In addition, GATK team provided best practices workflows
{kind=link}
- To help novice analysts
- To introduce workflows routinely used at ivory towers of research to rest of the world
- To standardize workflows across the labs (thus countries) to get reproducible results
Till recent times, GATK team provided workflows only DNAseq data, but not for RNAseq. However, GATK team came up best practices workflow for calling variants using RNAseq as outlined below:

(Based on workflow outlined here)
{kind=link}
In this note, following steps are executed and below the steps, a flow diagram is shown.
- Quality check by fastqc
- Trimming by cutadapt
- Alignment by tophat
- Alignment statistics collection by samtools
- Sorting aligned file by samtools
- Index the aligned file by samtools
- Deduplicating aligned file by picard
- Add Read Groups (RG) tag by picard
- Splitting N cigar reads by GATK
- Indel Recalibration (Creating intervals and Indel realignment with GATK)
- Recalibration files (First pass and Second pass with GATK)
- Quality check for recalibration (before and after plots by GATK)
- Base quality score recalibration (BQSR)
- Variant calling
- Variant filtering
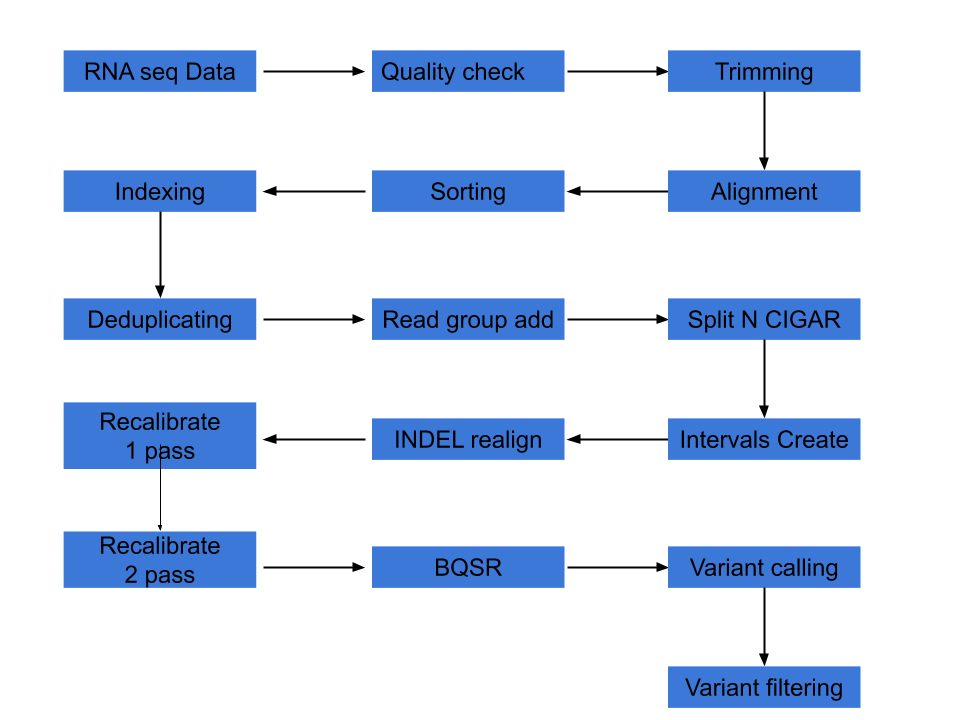 |
| Add caption |
RNA seq data for this note is downloaded from here and below is the bash script for running the pipeline: ( Please note that following additional steps are also included in the pipeline, but they are commented out. Do not mix variant calling (VCF) with genomic variant calling (gVCF))
- Discordant and concordant variant filtering
- gVCF calling
- merging gVCFs
######################
####bash script starts######
#####################
for i in $(ls ./raw_reads | grep ^[^d]| rev | cut -c 10- | rev | uniq)
do
# Make direcotry for fastqc
mkdir -p ${i%}_fastqc
# Run fastqc
fastqc -o ./${i%}_fastqc -f fastq /home/user/rnaseq/raw_reads/${i%}_R1.fastq /home/user/rnaseq/raw_reads/${i%}_R2.fastq
# Make directory to store cutadapt results
mkdir -p cutadapt
#Run cutadapt
cutadapt --quality-cutoff=20 --format=fastq -o ./cutadapt/${i%}_R1.cutadapt.fastq -p ./cutadapt/${i%}_R2.cutadapt.fastq /home/user/rnaseq/raw_reads/${i%}_R1.fastq /home/user/rnaseq/raw_reads/${i%}_R2.fastq
# Run top hat and save the out with directory extension _tophatout
tophat2 --no-coverage-search -o ./${i%}_tophat_out -G ./reference/genes_chr12.gtf -p 2 ./reference/chr12 ./cutadapt/${i%}_R1.cutadapt.fastq ./cutadapt/${i%}_R2.cutadapt.fastq
# Collect the stats
samtools flagstat ./${i%}_tophat_out/accepted_hits.bam > ./${i%}_tophat_out/accepted_hits.flagstats
#Sort bam
samtools sort -T /tmp/align.bam ./${i%}_tophat_out/accepted_hits.bam -o ./${i%}_tophat_out/accepted_hits.pos.sorted.bam
# Index the bam
samtools index ./${i%}_tophat_out/accepted_hits.pos.sorted.bam
# picard mkdir
mkdir -p ${i%}_picard
# Dedup bam
java -jar /opt/picard-tools-1.119/MarkDuplicates.jar \
METRICS_FILE=${i%}_picard/${i%}.dedup.metrics \
REMOVE_DUPLICATES=true ASSUME_SORTED=true \
VALIDATION_STRINGENCY=LENIENT \
CREATE_INDEX=true \
I=./${i%}_tophat_out/accepted_hits.pos.sorted.bam \
O=./${i%}_picard/q20.cutadapt.sorted.dedup.bam
# Add read groups
java -jar /opt/picard-tools-1.119/AddOrReplaceReadGroups.jar \
I=./${i%}_picard/q20.cutadapt.sorted.dedup.bam \
O=./${i%}_picard/q20.cutadapt.sorted.dedup.rg.bam \
CREATE_INDEX=true SO=coordinate RGID=${i%%_*} RGLB=${i%} RGPL=ILLUMINA RGSM=${i%} RGPU=GCCAAT
# mkdir gatk
mkdir -p ./${i%}_gatk
# Split N cigar reads
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T SplitNCigarReads \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_picard/q20.cutadapt.sorted.dedup.rg.bam \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.bam \
-U ALLOW_N_CIGAR_READS
#Sort bam
samtools sort -T /tmp/align.bam ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.bam -o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam
# Index the bam
samtools index ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam
# Create intervals
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T RealignerTargetCreator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam \
--known /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-L chr12 -o ./${i%}_gatk/forIndelRealigner.intervals
# Realign bam files
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T IndelRealigner \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam \
-known /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-L chr12 \
-targetIntervals ./${i%}_gatk/forIndelRealigner.intervals \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam
# Base recalibration First pass
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-knownSites /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-o ./${i%}_gatk/recal_data.table
# Base recalibration second pass
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-knownSites /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-L chr12 \
-knownSites /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-BQSR ./${i%}_gatk/recal_data.table \
-o ./${i%}_gatk/post_recal_data.table
# Create plots
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T AnalyzeCovariates \
-R /home/user/rnaseq/reference/chr12.fa \
-L chr12 \
-before ./${i%}_gatk/recal_data.table \
-after ./${i%}_gatk/post_recal_data.table \
-plots ./${i%}_gatk/recalibration_plots.pdf
# Apply BQSR to sequence
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T PrintReads \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-L chr12 \
-BQSR ./${i%}_gatk/recal_data.table \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam
# Variant calling
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./MeOH_REP1_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./MeOH_REP2_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./MeOH_REP3_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
--dbsnp /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-dontUseSoftClippedBases \
-stand_call_conf 20 \
-stand_emit_conf 20 \
-o ./meoh_output.raw.snps.indels.vcf
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./R3G_REP1_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./R3G_REP2_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./R3G_REP3_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
--dbsnp /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-dontUseSoftClippedBases \
-stand_call_conf 20 \
-stand_emit_conf 20 \
-o ./r3g_output.raw.snps.indels.vcf
# Discordant calls
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T SelectVariants \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./meoh_output.raw.snps.indels.vcf \
# --discordance ./r3g_output.raw.snps.indels.vcf \
# -o doutput.vcf \
# Concordant calls
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T SelectVariants \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./meoh_output.raw.snps.indels.vcf \
# --concordance ./r3g_output.raw.snps.indels.vcf \
# -o coutput.vcf \
# Variant filtering
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T VariantFiltration \
-R /home/user/rnaseq/reference/chr12.fa \
-V ./${i%}_gatk/output.raw.snps.indels.vcf \
-window 35 \
-cluster 3 \
-filterName FS \
-filter "FS > 30.0" \
-filterName QD \
-filter "QD < 2.0" \
-o ./${i%}_gatk/output.raw.snps.indels.filtered.vcf
#GenotypeGVCFs MeOH
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T GenotypeGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./MeOH_REP1_gatk/output.raw.snps.indels.g.vcf \
# -V ./MeOH_REP2_gatk/output.raw.snps.indels.g.vcf \
# -V ./MeOH_REP3_gatk/output.raw.snps.indels.g.vcf \
# -o meoh_output.vcf
#GenotypeGVCFs R3G
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T GenotypeGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./R3G_REP1_gatk/output.raw.snps.indels.g.vcf \
# -V ./R3G_REP2_gatk/output.raw.snps.indels.g.vcf \
# -V ./R3G_REP3_gatk/output.raw.snps.indels.g.vcf \
# -o r3g_output.vcf
#GenotypeGVCFs combine R3G Meoh
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T CombineGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V r3g_output.vcf \
# -V meoh_output.vcf \
# -o meoh_r3g_output.vcf
done
do
# Make direcotry for fastqc
mkdir -p ${i%}_fastqc
# Run fastqc
fastqc -o ./${i%}_fastqc -f fastq /home/user/rnaseq/raw_reads/${i%}_R1.fastq /home/user/rnaseq/raw_reads/${i%}_R2.fastq
# Make directory to store cutadapt results
mkdir -p cutadapt
#Run cutadapt
cutadapt --quality-cutoff=20 --format=fastq -o ./cutadapt/${i%}_R1.cutadapt.fastq -p ./cutadapt/${i%}_R2.cutadapt.fastq /home/user/rnaseq/raw_reads/${i%}_R1.fastq /home/user/rnaseq/raw_reads/${i%}_R2.fastq
# Run top hat and save the out with directory extension _tophatout
tophat2 --no-coverage-search -o ./${i%}_tophat_out -G ./reference/genes_chr12.gtf -p 2 ./reference/chr12 ./cutadapt/${i%}_R1.cutadapt.fastq ./cutadapt/${i%}_R2.cutadapt.fastq
# Collect the stats
samtools flagstat ./${i%}_tophat_out/accepted_hits.bam > ./${i%}_tophat_out/accepted_hits.flagstats
#Sort bam
samtools sort -T /tmp/align.bam ./${i%}_tophat_out/accepted_hits.bam -o ./${i%}_tophat_out/accepted_hits.pos.sorted.bam
# Index the bam
samtools index ./${i%}_tophat_out/accepted_hits.pos.sorted.bam
# picard mkdir
mkdir -p ${i%}_picard
# Dedup bam
java -jar /opt/picard-tools-1.119/MarkDuplicates.jar \
METRICS_FILE=${i%}_picard/${i%}.dedup.metrics \
REMOVE_DUPLICATES=true ASSUME_SORTED=true \
VALIDATION_STRINGENCY=LENIENT \
CREATE_INDEX=true \
I=./${i%}_tophat_out/accepted_hits.pos.sorted.bam \
O=./${i%}_picard/q20.cutadapt.sorted.dedup.bam
# Add read groups
java -jar /opt/picard-tools-1.119/AddOrReplaceReadGroups.jar \
I=./${i%}_picard/q20.cutadapt.sorted.dedup.bam \
O=./${i%}_picard/q20.cutadapt.sorted.dedup.rg.bam \
CREATE_INDEX=true SO=coordinate RGID=${i%%_*} RGLB=${i%} RGPL=ILLUMINA RGSM=${i%} RGPU=GCCAAT
# mkdir gatk
mkdir -p ./${i%}_gatk
# Split N cigar reads
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T SplitNCigarReads \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_picard/q20.cutadapt.sorted.dedup.rg.bam \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.bam \
-U ALLOW_N_CIGAR_READS
#Sort bam
samtools sort -T /tmp/align.bam ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.bam -o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam
# Index the bam
samtools index ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam
# Create intervals
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T RealignerTargetCreator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam \
--known /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-L chr12 -o ./${i%}_gatk/forIndelRealigner.intervals
# Realign bam files
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T IndelRealigner \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.bam \
-known /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-L chr12 \
-targetIntervals ./${i%}_gatk/forIndelRealigner.intervals \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam
# Base recalibration First pass
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-knownSites /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-o ./${i%}_gatk/recal_data.table
# Base recalibration second pass
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T BaseRecalibrator \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-knownSites /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-L chr12 \
-knownSites /home/user/ngs_exercise/data/hg19/Mills_and_1000G_gold_standard.indels.hg19.chr12.vcf \
-BQSR ./${i%}_gatk/recal_data.table \
-o ./${i%}_gatk/post_recal_data.table
# Create plots
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T AnalyzeCovariates \
-R /home/user/rnaseq/reference/chr12.fa \
-L chr12 \
-before ./${i%}_gatk/recal_data.table \
-after ./${i%}_gatk/post_recal_data.table \
-plots ./${i%}_gatk/recalibration_plots.pdf
# Apply BQSR to sequence
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T PrintReads \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.bam \
-L chr12 \
-BQSR ./${i%}_gatk/recal_data.table \
-o ./${i%}_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam
# Variant calling
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./MeOH_REP1_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./MeOH_REP2_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./MeOH_REP3_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
--dbsnp /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-dontUseSoftClippedBases \
-stand_call_conf 20 \
-stand_emit_conf 20 \
-o ./meoh_output.raw.snps.indels.vcf
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T HaplotypeCaller \
-R /home/user/rnaseq/reference/chr12.fa \
-I ./R3G_REP1_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./R3G_REP2_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
-I ./R3G_REP3_gatk/q20.cutadapt.sorted.dedup.snc.sorted.realigned.recal.bam \
--dbsnp /home/user/rnaseq/reference/chr12.dbsnp.b141.b37.hg19.tidy.vcf \
-dontUseSoftClippedBases \
-stand_call_conf 20 \
-stand_emit_conf 20 \
-o ./r3g_output.raw.snps.indels.vcf
# Discordant calls
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T SelectVariants \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./meoh_output.raw.snps.indels.vcf \
# --discordance ./r3g_output.raw.snps.indels.vcf \
# -o doutput.vcf \
# Concordant calls
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T SelectVariants \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./meoh_output.raw.snps.indels.vcf \
# --concordance ./r3g_output.raw.snps.indels.vcf \
# -o coutput.vcf \
# Variant filtering
java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
-T VariantFiltration \
-R /home/user/rnaseq/reference/chr12.fa \
-V ./${i%}_gatk/output.raw.snps.indels.vcf \
-window 35 \
-cluster 3 \
-filterName FS \
-filter "FS > 30.0" \
-filterName QD \
-filter "QD < 2.0" \
-o ./${i%}_gatk/output.raw.snps.indels.filtered.vcf
#GenotypeGVCFs MeOH
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T GenotypeGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./MeOH_REP1_gatk/output.raw.snps.indels.g.vcf \
# -V ./MeOH_REP2_gatk/output.raw.snps.indels.g.vcf \
# -V ./MeOH_REP3_gatk/output.raw.snps.indels.g.vcf \
# -o meoh_output.vcf
#GenotypeGVCFs R3G
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T GenotypeGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V ./R3G_REP1_gatk/output.raw.snps.indels.g.vcf \
# -V ./R3G_REP2_gatk/output.raw.snps.indels.g.vcf \
# -V ./R3G_REP3_gatk/output.raw.snps.indels.g.vcf \
# -o r3g_output.vcf
#GenotypeGVCFs combine R3G Meoh
#java -jar /opt/gatk-3.4-46/GenomeAnalysisTK.jar \
# -T CombineGVCFs \
# -R /home/user/rnaseq/reference/chr12.fa \
# -V r3g_output.vcf \
# -V meoh_output.vcf \
# -o meoh_r3g_output.vcf
done
##############################
###bash script ends##############
####################